FGFR1 amplification

I am asking for collective wisdom for those that have this genetic mutation. FGFR1 amplification is reported to be in 10-27% in breast cancer (per FoundationOne).
I progressed after 3+ great years on Ibrance/Letrozole. Recent liver biopsy report from Foundation One(F1) showed FGFR1 amplification and ATM inversion exon 55.
Currently on Xeloda (capecitabine).
Are my antiestrogen days over, to include future oral SERDS? Can I still take Verzenio as a monotherapy? Anyone have success on Afinitor with this mutation?
Any successful clinical trials with FGFR1 inhibitors?
F1 suggested for FGFR1 amp: NCT03344536 (Debio1347), NCT04233567 (Infigratinib), NCT02549937 (Sulfatinib).
Comments
-
FGFR1 amplifications are a common route to endocrine resistance, so hopefully people with that mutation will chime in and say what worked for them.
here is one review-but mentions some work saying that FGFRs often do not drive growth in these cancers? And that may not be a big loss, given all the side effects the FGFR inhibitors seem to have, tho at least this is an active area of development
https://www.onclive.com/publications/oncology-live...
"In vitro and sequencing studies suggest that FGFR is not a dominant oncogene in FGFR1-amplified cancers, whereas FGFR2-amplified cells are highly addicted to FGFR signaling and demonstrate an apoptotic response to FGFR inhibition"
0 -
Thanks Cure-ious. I was really hoping my bx was Pik3 positive or ESRI positive as I knew there were treatments to use. But now I feel I will go the route of TNBC with nonhormonal treatments. Which there are plenty of success stories.
0 -
nI was just reading that lung cancers only about 10% of FGFR1-amplified cancers responded to FGFR inhibitors, ubt did better with combinations that included MET or MEK inhibitors
There are other targeted drugs too- HDAC inhibitors, BET inhibitors, etc, but check if FGFR1-amplified cancers are sensitive or not..
0 -
SandiBeach - I have FGFR1. I did have six months of success on Affinator after Ibrance. Although some do struggle with Affinitor, I was fortunate that It was a very easy drug for me. Verzenio didn't work for me after Ibrance although it does for some. Also did a short time on an FGFR trial of erdafitinib, Faslodex and Ibrance (second time for me on both Fas and Ibrance). However, due to side effects and large rise in markers, my onc recommended that I leave the trial. I've been on Abraxane since. Doing well. Good luck with your research.
0 -
Hi Moissy,
I have read so much about FGFR1 amplification that I think my head is going to explode.
Thank you for sharing your information. It is comforting to hear from others.
From what I read, Afinitor (mTOR) seems to allow the antiestrogen to work if you have the FGFR1 amp gene. Which is odd as this gene is suppose to cause resistance to all antihormonal therapy.
There was one study (I will try to link the study with an edit) on a patient who"s Ibrance/ Letrozole stopped working after 11 months. Biopsy showed FGFR1 amp gene mutation. She enrolled in a clinical trial ( still can"t find the old trial#..). The trial was antiestrigen + CK4/6 inhibitor AND Afinitor (Everolimus). She responded for 14 months and was still doing fine when article was published July 2018.
So basically, they were studying antihormonal + CK4/6 inhibitor + mTOR to work around the FGFR1 ampl gene.
Question: Does anyone know if you have a subsequent progression and the new biopsy shows more mutations, does that mean the current mutation is no longer the pathway?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC68255...
If someone could better understand this article, I would be grateful. The trial must have been a private sponsored trial as I could not find the reference in the article. I think it also mentioned that Fulvestrant + Afinitor had better response than Fulv + Afin + Versenio. I could have misinterprited, so beg forgiveness of ignorance in advance.
0 -
Sandi- That's the problem, whether its a pre-existing mutation or one that popped up in the resistant cancer, no test is going to know which ir any of these mutations is driving the growth of the cancer. Actually that is where the oncologists experience with many patients, or this board, can be so helpful of knowing what are real-world experiences.FGFR1 amplifications are better studied in lung cancer, and most of those cases its other pathways, like mTOR, that are driving the growth, and FGFR is just a bystander mutation not doing much.
So, just be looking for people like Moissy, we must have many more on this site, but because testing is relatively new a lot of people may not know they have the mutation
Hoping more people weigh in on their experiences..
0 -
Hi Cure-ious. Yes, you are right. Since I did not have the F1 done with original liver bx in 2016, I do not know if I had the mutation at met dx. But I did get a good rin on I/L...39 months. Did it stop because of FGFR1 amp..I don't know.
I was expecting the ESR1 or PIK3 mutation (both negative), so was just taken off guard. My poor MO will be bombarded with questions next week.
0 -
Did some editing to my post with attached article..
0 -
Unfortunately, none of this stuff is black and white. The only thing that we know for sure about cancer is that it is unpredictable. Logic about which treatments should work and why doesn’t always follow.
As you know, I did a trial of erdafitinib (Balversa, now approved for urothelial cancer). It didn’t work as mono therapy and caused some scary side effects. I would try it again in combination though, with the erdafitinib at a lower dose.
I also have the PIC3K mutation and tried Afinitor for 3 months. It did not reverse the cancer growth but did slow it down a bit. I wonder it it would have worked if I gave it more time. I have heard it is a slow acting drug and takes time to start to work.In my humble opinion, the best way to view genetic profile results are as “clues”. They are worth investigating and some may lead to treatments that work but many may end up being dead ends. Cancer doesn’t follow logic.
0 -
Thank you JFL.
This is exactly the info that I am looking for-real world experiences with these drugs and targeting mutations. Seems like it will always be a gamble on what really works.
I see my MO next week and looking forward to hearing what she has actually seen with regards to mutations vs tx choices.
0 -
I thought this was an interesting trial, combines immunotherapy + PARP inhibitor + Faslodex. You have to have a mutation in BRCA or other genes that are involved in DNA repair, including FGFR mutations:
Deleterious germline or somatic alterations implicated in the HRR pathway (ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FAND2, FANCL, MRE11A, NBN, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D and RAD54L) or in MSI status or other actionable genes (AKT1, ESR1, FGFR1, FGFR2, FGFR3, and PIK3CA) all based on central tumor next generation DNA sequencing performed at screening visit.
Unfortunately offered only in MontPelier France, but if they get good results it will surely get picked up more broadly
https://clinicaltrials.gov/ct2/show/NCT04053322#co...
0 -
Thank you Cure-ious for that info.
I bookmarked the below trial that is anti FGFR1 amp (anti tyrosine).
NCT03238196, Erdafitinib, Fulvestrant, Ibrance. I think the CA location is the only one enrolling..but might be on hold during COVID19.
Also looking at mTOR combos that are being looked at for FGFR1 amp.
My MO said she would probably put me on Aromasin and Afinitor next , the mTOR inhibitor. From what I read, a trio combo of antiestrogen + mTOR+ antiFGFR can overcome the antiestrogen resistance. I have so many articles bookmarked..I might be getting confused.
My MO said she would follow standard of care with future treatments as we don't know 100% certainty what drives treatment resistance even with noted mutations.
The trials would have to be timed perfectly as you have to be off tx and if you have aggressive cancer, esp liver mets, that could not be in my best interest.
I will have to reread my notes and print out the pertinent ones so I am repeating accurate info.
My life would be a tad easier if I had the straight forward ESR1 or PIK3 mutations.
0 -
I want to ask questions to those who understand mutations that cause resistance to antihormonals.
1. Once a biopsy identifies a mutation in the tumor, is this mutation present forever or can it disappear and become a nonissue?
2. Is the mutation always actionable..meaning does it inhibit antiestrogen therapy forever? or will the resistant pathway stop when a new mutation occurs?
3. Will the mutation eventually allow tumor to resensitize to antiestrogen therapy?
4. If you are off antiestrogen therapy, can you still acquire mutations, such as ESRI ot PIC3?
5. Has anyone had success going back on antiestrogens while harboring mutations that have no actionable treatment at this time?
I realize these might be silly questions, just not sure how to find a geneticist who specializes in acquired breast cancer mutations. Maybe I will call Foundation One.
0 -
These are all good questions, and ones I am not qualified to answer! I have had informative conversations with folks at F1 and Guardant about my results. To get to the scientists who know stuff, you have to ask questions that throw the call screeners and convince them you have legitimate questions requiring expert responses. As always, you can’t exactly ask for medical advice, but you can ask about interpretation and general science, and how to best use their tests.
0 -
Before retiring I worked in a breast cancer research lab focused on studying the development of cancer cell resistance to taxol and anthracyclines. This is how I would answer your questions:
1. Once a biopsy identifies a mutation in the tumor, is this mutation present forever or can it disappear and become a nonissue? A germ line mutation (ie BRCA, ATM or FGFR2 in my case) or somatic mutation (in checkpoint inhibitors) is likely what is driving the original tumor growth and will be there as long as there is original tumor. An acquired mutation (in ESR or growth factor amplification) as a result of treatment and can result in hormone resistance, is also permanent. But one should keep in mind that tumors are dynamic. Some cells express mutations and others might not. Some are responding to antihormones and cyclin inhibitors, others aren't, or, with time, can develop resistance. As in our studies, there was always a very small subpopulation that developed resistance to taxol. And, hopefully, a healthy immune system would take care of those cells. If not, a new tumor would develop now resistant to taxanes that required new treatment.
2. Is the mutation always actionable..meaning does it inhibit antiestrogen therapy forever? or will the resistant pathway stop when a new mutation occurs? The mutation is always actionable as long as it helps the tumor to survive. If cancer treatment drugs are given for a long enough time they may cause NED and/or a subpopulation will acquire a new mutation that will supersede the treatment and allow the surviving cells to continue to grow. I would surmise the surviving cells have the original and a new mutation based on our studies. But I don't know that for a fact with antihormones.
3. Will the mutation eventually allow tumor to resensitize to antiestrogen therapy? Mutations are acquired by tumor cells to help them survive. If a mutation developed in response to antiestrogens, which is common, it was to help tumor cells survive in the presence of antihormones, likely by usurping the ER pathway. Fortunately, there are cancer drugs available to help negate the effect of the new mutation (ie, PI3K inhbitors)
4. If you are off antiestrogen therapy, can you still acquire mutations, such as ESRI ot PIC3? I think this depends on what is the driving force for the tumor growth. Chances are that if tumor growth continued during antihormone treatment, other growth factors may be at work (ie, EGF, FGF, IGF) and/or mediators of their pathway(s) may be constitutively expressed (PI3K, mTOR). That you were PR- like me, suggests that there was overexpression of a growth factor that down-regulated PR, or that our ER function was somehow compromised. In either case, this may be why PR- individuals are less likely to respond well to antihormonals.
5. Has anyone had success going back on antiestrogens while harboring mutations that have no actionable treatment at this time? On this site, I've seen that docs have switched around TAM and different AIs with CDK inhibitors, everolimus, SERDs et al, per protocol. If new tumor (not metastasis) develops, seems antihormones could be given again if it, too, is ER+.
0 -
Hello Murfy, I was greatly honored that you responded to each of my questions. Thank you.
It is with much appreciation that you used your background professional knowledge to help me understand the basics of genetic mutations. I did not know where to start.
Since my recent liver bx showed two actionable mutations, just assume no one really knows which one is driving the progression..or neither of them.
My MO is thinking of Afinitor next as it is mTOR inhib, plus antiestrogen. But personally, don't have much confidence it will work. I guess when Xeloda stops working, I need a second opinion. It is just sometimes, I feel I will be fighting against any time delay.
Thank you again.
0 -
Murfy. Thanks for sharing your knowledge!
I was not aware that FGFR2 can suppress expression of PR..
How did you learn you have an FGFR2 mutation?
We were discussing a report showing that FGFR2-hi cancers respond well to ibrance, opposite to cancers with FGFR1-amplification, do you know why that might be?
eg, what other differences between these pathways?
do both induce cyclin D? thanks
0 -
Sandi, if, based on your signature, I could be so bold as to posit the following, as a non-clinician: A germ-line mutation in the DNA-repair mechanism gene ATM could have been involved in the initiation of your breast cancer. Is that what you are thinking? Later, an acquired mutation causing FGFR1-amp might be driving your current tumor growth. That you have residual PR+ suggests some functional ER may also be contributing to tumor cell proliferation. Your current treatment inhibits rapidly dividing cell proliferation and could cause NED. If not, an mTOR inhibitor might do so. I read a recent Nature Comm paper of pre-clinical study combining AI+checkpoint inhibitor+ FGFR inhibitor and this combined treatment inhibited FGFR1-amplified tumor growth. I noticed clinical trials including several different FGFR inhibitors. Have you considered a clinical trial as a future option?
0 -
Hi Murfy, you make me smile as I have been researching the same clinical trials, that is, an antiestrogen + targeted inhibitor + antiFGFR.
When the liver mets were diagnosed in 2016, my MO sent my blood to check for germline mutations. There were none. She did not order a F1 on my bx in 2016 as it was not going to change her treatment choice..AC to kill aggressive cells. She also felt if I waited for a future F1, the company would find more mutations. The biologist in me wished we had sent it. I think I still have the blocks.
The recent F1 report gave suggested trials for each mutation, but also discussed actionable tx with a PARP inhib due to the ATM. It just confuses me even more as to what is driving progression..ATM, FGFR1 amp or some unknown force? And really what do I take when progression happens again?
My readings so far are leaning toward mTOR inhib(Afinitor), but confused as you have to combine with antiestrogen..which apparently isn't working anymore. Oh..how I wish sadly for the PK3 ot ESRI mutations.
My MO said that F1 will update us when new trials, meds become available in future. I just have to be careful not to get on tx that would disquailfy me from a trial of interest. My MO is also concerned with aggressive disease, that waiting for washout for trial could be dangerous.
Ok..rambling. Sorry.
0 -
Hi Cure-ious. I enjoy reading your posts and the detailed science within. When some of my family members began to get an unusual sequelae of illnesses, I sequenced my own DNA in the lab and then uploaded raw DNA into an online, comprehensive program that identifies health-related SNPs. An interestingly depressing list of genes was identified for numerous health conditions (this program is definitely not for everyone!). At the top of my breast cancer list (which was small) was a triple mutation in my FGFR2 gene each/all of which predispose to breast cancer typically in post-menopausal women.
Trained in endocrinology, my PR- status interested me, especially as I was almost 100% ER+ and ER is responsible for maintaining a healthy population of PR. Many papers later, I deduced that either the mutation caused degradation of PR and ER ran amok or that ER signaling was compromised. A 2019 review paper (link below) explained how the latter can be caused by the triple mutation. However, other growth factor malfunctions, notably HER2 and EGF/FGF amplifications or mutations, can alter ER signaling and/or degrade PR, and itself drive proliferation.
What is FGFR2-hi?
https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1236-6
0 -
Murfy,
Below is the graph I am trying to understand. In part A, it shows that people who take Ibrance and AI and have low PD1 levels (dashed blue line) have a longer PFS response interval than those with high PD1 expression (solid blue line), and PD1 levels had no significant effect on the control group, which were not taking Ibrance (red lines)
In part B, it shows a better PFS interval for those with FGFR2-high expression (solid blue line at top; presumably due to mutation or amplification) when taking Ibrance, whereas FGFR2 normal did not do as well (dashed blue line) and FGFR2 levels had no effect in the control group not getting Ibrance (red)
In part C it shows the same graph for high ERBB3 (HER3), which also correlated with a stronger response to Ibrance specifically.
So we know FGFR1 amplifications pre-dispose to progression and endocrine resistance, however these data indicate that FGFR2-hi has the opposite effect, it specifically makes response to Ibrance better, and the papers I've read treat FGFR1 and FGFR2 mutations as interchangeable, do not provide a possible mechanism that could explain their opposing responses to Ibrance.
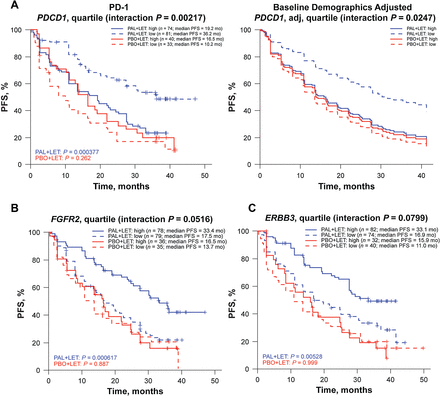 0
0 -
BTW, the PD1 levels have a remarkable effect as well- if you have high PD1 expression, you are not getting much of any benefit from Ibrance, the curves for those cancers are very similar to the responses seen with letrozole alone, just a couple of months benefit.
There are some very involved and direct interactions between the CDK4,6 and PD1 pathways, including but not limited to the fact that CDK4 promotes degradation of PD1, so drugs like Ibrance can increase PD1 and are reported to improve response to immunotherapy, although so far we haven't heard great validation of that in the ongoing clinical trials.
But what is the reason explaining the loss of benefit from Ibrance at high PD1 expression? And would inhibiting PD1 activity (by Keytruda) cause these cancers to become sensitive to Ibrance-Femara?
0 -
Hi Cure-ious. My bx was negative for PDL1..maybe that is why I lasted so long on I/L. I guess we are assuming it was the acquired FGFR1 amp that drove progression..but who really knows. It just the likely culprit.
I just assumed I was going to have the ESRI or Pik3 mutation. I had my treatment all lined up..kind of sad/funny that you really can't plan ahead. But you can do your research!
My MO wants me to try A/A with next progression as mTOR seems to have moderate response to tumors with this mutation. Somewhere I have that source. This is when I will need a 2nd opinion.
Thank you for contributing your research to this thread. Maybe these conversations will help someone out there who has the same questions.
S
0 -
Sandi- I bet that's right, you were flying along that top curve (where it looks like about half of those made it out 50 months) until the fgfr1 amplification happened
its well worth a second opinion to ask about an FGFR inhibitor combination with ibrance-letrozole, maybe you can respond again for a long time, and the NEXT progression might happen happen via esr1 or pi3k mutation...
What's not clear is if that combo would be of any help for a fgfr2 mutation? because it looks like one would lose ibrance sensitivity
0 -
Another question is, lets say they had an opening in that anti-fgfr trial, but you are stable and doing fine on xeloda-do you wait for progression, or try the trial knowing that you can jump back to xeloda if it doesn't work? I know oncologists always do drugs sequentially, but if you jumped before progression the cancer state is presumably unchanged- following progression on xeloda, would it still respond?
these are academic questions and I imagine no way of knowing; it would be great to have something you know will work in your back pocket, but this is why all this stuff is a crapshoot; well its a question for the second opinion, or somebody running the trial, really...
0 -
Here's my take...
I'm assuming all are ER/PR+ and treatment started upon met diagnosis.
PD1 data indicates that Ibrance most effective when PD1 low. High PD1/L seems to be associated with high cytokines and pro-inflammatory mediators in tumor microenvironment.
Moreover, hi-FGFR2 and hi-ERBB3 are associated with lower levels of pro-inflammatory IL-6 (and probably other cytokine) levels in breast epithelium.
So, a reduced inflammatory environment, as occurs when PD1 is low and FGFR2 or ERBB3 are high makes Ibrance-AI combo more efficacious. High levels of IL-6 increase cancer cell proliferation via JAK/STAT at G1-S and may 'over-ride' ability of therapeutic dose of Ibrance to inhibit at G1-S.
Wow, thanks for making me exercise my brain!
0 -
crossposting from Afinitor thread:
Hi ladies,
After 7 rounds of Kisqali/Fulvestrant, it looks like I might have progression and will be on my way to Afinitor. I went all the way into the normal range with TM's but my tumor markers are rising again. I have the FGFR1 amplification (lucky me) and have scans coming up to verify progression. I'm not thrilled, but I'm also not surprised as I know the FGFR makes me resistant to Kisqali, so I'm glad for what I got out of it. I was hoping for 12, expected 6, got 7, so a little win. I'm guessing I'll stay on Fulvestrant as well, but not sure yet.
I also don't know if I should try a clinical trial first or wait until after Afinitor? My MO isn't the most receptive to new ideas, I need to find another one I click with a little better, but now doesn't seem like the time to go doctor shopping. Maybe a second opinion at MD Anderson would be a better move. From what I have read and researched (mostly from the amazing researchers on this board) Afinitor is the logical next step, so I'm not in a huge hurry.
I've been feeling so good on Kisqali, I've been able to push the cancer out of my mind, but with Afinitor, I'm afraid the side effects will keep it front and center. That's almost as bad as the SE's themselves. Maybe I'll get lucky again, who knows. Anyway, I'm expecting (hoping) for good results on Afinitor - at least 2 years, hopefully 3. Am I being too optimistic? I really hope not.
Sorry for the stream of consciousness verbal vomit post, but I'm trying to process this new info. Thanks for listening.
0 -
LaureninPHX, from what I read with this mutation (still looking for my source), AA has some good response due to mTOR inhinitor..different pathway than CK4/6.
It would be even better if combined with antiFGFR. But no luck in finding trial. There is a trial at MD Anderson that I want to explore..antiFGFR + SERD. Fulvestrant use excludes you.
I am meeting with my MO in 2 weeks after liver MRI to discuss options. If stable..do I continue with Xeloda or go to trial and hold Xeloda for later?
Keep us posted. We all learn from each other.
0 -
Thank you, Sandi - Good luck with your MO/MRI - hoping for the best.
I'm glad this thread is here, I've already learned so much and I'll definitely keep everyone up to date.
0 -
I think this is the article that I was referencing... November 2019
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6825550/
Suspect that is why my MO talked about Aromosin/Afinitor next.
0

